Bayesian Transmission Modeling
Source:vignettes/bayesian-transmission.Rmd
bayesian-transmission.RmdIntroduction
This package provides a Bayesian framework for transmission modeling on an individual patient level. Modeling is conducted through Markov Chain Monte Carlo (MCMC) methods.This document will explain the basic usage of the package, specification of parameters, and the output of the model.
Data Structure
The algorithms expect a longitudinal data set with the following
columns: * facility: The facility where the event occurred.
* unit: The unit within the facility where the event
occurred. * time: The time at which the event occurred. *
patient: The patient involved in the event. *
type: The type of event.
The package includes a simulated dataset,
simulated.data.
pillar::glimpse(simulated.data)
#> Rows: 8,360
#> Columns: 5
#> $ facility <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
#> $ unit <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1…
#> $ time <dbl> 0.060978, 0.061978, 1.560978, 2.883323, 2.884323, 1.422631, 1…
#> $ patient <int> 1, 1, 1, 1, 1, 10, 10, 10, 10, 19, 19, 19, 23, 23, 23, 28, 28…
#> $ type <int> 0, 1, 10, 1, 3, 0, 1, 1, 3, 0, 1, 3, 0, 1, 3, 0, 1, 1, 3, 0, …There are 12 different types of events that can be specified in the
type column. These are, expected numerical codes shown in
parentheses:
- Admission (0) and Discharge (3)
- Surveillance Testing Results
- Negative Test (1)
- Positive Test (2)
- Clinical Testing Results
- Negative Test (4)
- Positive Test (5)
- Generic Testing
- Negative Test (7)
- Positive Test (8)
- Antibiotic Use
- single dose (9)
- Start (10)
- Stop (11)
- Isolation Procedures
- Start (6)
- Stop (7)
Not all events need to be used in every data set, but the model
selected should reflect the data that is available. Care should be taken
to correctly code the data. The EventToCode and
CodeToEvent functions can be used to convert between.
table(CodeToEvent(simulated.data$type))
#>
#> abxoff abxon admission discharge negsurvtest possurvtest
#> 297 725 2183 2183 2749 223Model Specification
Model Choice
Since the model is implemented in C++ for speed and efficiency, only the specified models can be used. The currently implemented models are:
-
"LinearAbxModel", A Linear model with antibiotic use as a covariate. -
"MixedModel", and "LogNormalModel"
Model specification and all parameters are controlled through
constructor functions of the same name, or generically through the
LogNormalModelParams() function.
For all models there is the choice of either a 2 state (susceptible
and colonized) or 3 state (susceptible, colonized, and recovered or
latent) model. Number of states is set through the nstates
parameter, and the number of states in the model overrides what may be
specified in any individual component.
Parameters
The remainder of the parameters are grouped into the following categories:
-
Abx, Antibiotic use, -
AbxRate, Antibiotic rates, -
InUnit, In unit infection rates, -
OutOfUnitInfection, Out of unit infection rates, -
Insitu, In situ parameters, - Testing:
-
SurveilenceTest, Surveillance testing, -
ClinicalTest, Clinical testing.
-
Unless otherwise specified the parameters are all distributed gamma with specified shape and rate parameters. Each parameter can also be left as fixed or be sampled at each iteration of the MCMC.
Specifying parameters
Parameters for the model may be specified by the Param()
function. This function takes up to four arguments:
1. `init`, is the initial value of the parameter.
2. `weight`, is the weight of the prior distribution in updates.
3. `update`, a flag of if the parameter should be sampled in the MCMC algorithm.
`FALSE` indicates that the parameter should be fixed, and is by default `TRUE` when `weight` is greater than zero.
4. `prior`, the mean of the prior distribution. Taken with the weight will fully parameterize the distribution.Important: Always explicitly specify
init values for parameters to ensure reproducibility and
avoid potential numerical issues.
# Fully specified parameter.
Param(init = 0, weight = 1, update = TRUE, prior = 0.5)
# Fixed parameter with explicit init
# Weight = 0 implies update=FALSE and prior is ignored.
Param(init = 0, weight = 0)
# Updated parameter that starts at zero.
Param(init = 0, weight = 1, update = TRUE)
# Short form for fixed parameter
Param(init = 0, weight = 0)
Abx Antibiotic use
Antibiotic use is specified by the Abx parameter. This
parameter is a list constructed with the AbxParams()
function with the following components:
-
onoff, If antibiotics are being used or not. The two following parameters are only used ifonoffisTRUE. -
delay, the delay for the antibiotic to take effect. -
life, the duration where the antibiotic to be effective.
abx <- AbxParams(onoff = 0, delay = 0.0, life = 2.0)Currently, all antibiotics are assumed to be equally effective and
have the same duration of effectiveness. Note: onoff = 0
means antibiotics are turned off for this example (set to 1 to
enable).
AbxRate Antibiotic rates
The AbxRate parameter control the antibiotic
administration rates.
abxrate <- AbxRateParams(
# Fixed parameters when antibiotics are off
uncolonized = Param(init = 1.0, weight = 0),
colonized = Param(init = 1.0, weight = 0)
)Here since both parameters are non-zero both will be updated. A rate
of zero for either would indicate that group would never be on
antibiotics. When antibiotics are turned off
(Abx$onoff = 0), these parameters are typically fixed
(weight = 0).
InUnit In unit infection rate
Transmission within unit is the main defining characteristic that
differentiates models. For example the linear antibiotic model,
LinearAbxModel(), is differentiated from the log normal
model, LogNormalModelParams() by the use of a
ABXInUnitParams() for the InUnit argument
rather than the LogNormalInUnitAcquisition() which does not
take into account antibiotic use. All in unit transmission is defined in
terms of acquisition, progression, and clearance.
Acquisition Model
In the base log normal antibiotic model,
LogNormalABXInUnitParameters() log acquisition probability
at time
is a linear function.
Where represents the coefficient corresponding to the amounts, represents the total number of colonized patients () at time , the number of colonized on antibiotics (), and and indicate susceptible () patients currently () or ever () on antibiotics.
The linear antibiotic (LinearAbxAcquisitionParams) takes
a more complicated form for the acquisition model.
acquisition <- LinearAbxAcquisitionParams(
base = Param(init = 0.001, weight = 1), #< Base acquisition rate (Updated)
time = Param(init = 1.0, weight = 0), #< Time effect (Fixed)
mass = Param(init = 1.0, weight = 1), #< Mass Mixing (Updated)
freq = Param(init = 1.0, weight = 1), #< Frequency/Density effect (Updated)
col_abx = Param(init = 1.0, weight = 0), #< Colonized on antibiotics (Fixed)
suss_abx = Param(init = 1.0, weight = 0), #< Susceptible on antibiotics (Fixed)
suss_ever = Param(init = 1.0, weight = 0) #< Ever on antibiotics (Fixed)
)Progression Model
In the 3 state model there is a latent state () and the progression model controls how patient transition out of the latent state. The base rate can be affected by currently being on antibiotics or ever being on antibiotics.
the linear antibiotic model is:
Where here we use for the coefficients, but the notation is the same.
progression <- ProgressionParams(
rate = Param(init = 0.0, weight = 0), #< Base progression rate (Fixed for 2-state)
abx = Param(init = 1.0, weight = 0), #< Currently on antibiotics (Fixed)
ever_abx = Param(init = 1.0, weight = 0) #< Ever on antibiotics (Fixed)
)Clearance Model
The clearance model is the same as the progression model in both the log normal and the linear cases, the coefficients however are independent.
clearance <- ClearanceParams(
rate = Param(init = 0.01, weight = 1), #< Base clearance rate (Updated)
abx = Param(init = 1.0, weight = 0), #< Currently on antibiotics (Fixed)
ever_abx = Param(init = 1.0, weight = 0) #< Ever on antibiotics (Fixed)
)
inunit <- ABXInUnitParams(
acquisition = acquisition,
progression = progression,
clearance = clearance
)Out of Unit Importation
The out of unit parameters control the rate at which admissions come in, and which state they enter in.
outcol <- OutOfUnitInfectionParams(
acquisition = Param(init = 0.001, weight = 1),
clearance = Param(init = 0.01, weight = 0),
progression = Param(init = 0.0, weight = 0)
)In Situ
I’m not sure what these parameters do. It’s a set of gamma distributed parameters one for each state. The updates and probabilities are not time dependent.
When updating the rates for each state are sampled from a distribution. Then all three are normalized to sum to 1.
insitu <- InsituParams(
# Starting 90/10 split uncolonized to colonized
# For 2-state model, latent probability is 0
probs = c(uncolonized = 0.90,
latent = 0.0,
colonized = 0.10),
# Prior values for Bayesian updating
priors = c(1, 1, 1),
# Which states to update (latent is fixed at 0 for 2-state model)
doit = c(TRUE, FALSE, TRUE)
)Testing
There are two types of testing, surveillance, which is conducted routinely at regular intervals such as on admission then every 3 days after, and clinical, where the testing is precipitated by staff, and thus the timing is informative.
Surveillance Testing
The timing of surveillance testing is assumed to not be informative. Therefore, surveillance testing is only parameterized in terms of probability of a positive test given the underlying status. Surveillance test parameters are updated with a sample from a distribution where and are the number of positive and negative tests respectively for state .
surv <- SurveillanceTestParams(
# Probability of a positive test when uncolonized (false positive rate)
# IMPORTANT: Must be > 0 to avoid -Inf likelihood. Use small value like 1e-10.
# Setting to exactly 0.0 causes log(0) = -Inf if any uncolonized patient tests positive.
uncolonized = Param(init = 1e-10, weight = 0),
# Probability of a positive test when colonized (true positive rate/sensitivity)
# Starting at 0.8, will be updated during MCMC
colonized = Param(init = 0.8, weight = 1),
# Latent state (for 2-state model, this is not used but must be specified)
latent = Param(init = 0.0, weight = 0)
)Clinical Testing
Since clinical testing time is informative, clinical testing is assumed to be at random within infection stage. The rate of testing within each stage is sampled from a gamma distribution. Sensitivity/Specificity are handled the same as surveillance testing and the likelihood is multiplicative between rate and effectiveness.
clin <- RandomTestParams(
# Rate of testing when uncolonized
uncolonized = ParamWRate(
param = Param(init = 0.5, weight = 0),
rate = Param(init = 1.0, weight = 0)
),
# Rate of testing when colonized
colonized = ParamWRate(
param = Param(init = 0.5, weight = 0),
rate = Param(init = 1.0, weight = 0)
),
# Latent state (for 2-state model, not used but must be specified)
latent = ParamWRate(
param = Param(init = 0.5, weight = 0),
rate = Param(init = 1.0, weight = 0)
)
)All Together
params <- LinearAbxModel(
nstates = 2,
Insitu = insitu,
SurveillanceTest = surv,
ClinicalTest = clin,
OutOfUnitInfection = outcol,
InUnit = inunit,
Abx = abx,
AbxRate = abxrate
)Running the Model
The model is run through the runMCMC() function. The
function takes the following arguments:
-
data: The data frame with patient event data -
modelParameters: The model specification (created above) -
nsims: Number of MCMC samples to collect after burn-in -
nburn: Number of burn-in iterations (default: 100) -
outputparam: Whether to save parameter values at each iteration (default: TRUE) -
outputfinal: Whether to save the final model state (default: FALSE) -
verbose: Whether to print progress messages (default: FALSE)
system.time(
results <- runMCMC(
data = simulated.data_sorted,
modelParameters = params,
nsims = 100,
nburn = 10,
outputparam = TRUE,
outputfinal = TRUE,
verbose = FALSE
)
)
#> user system elapsed
#> 17.761 16.326 17.056Analyzing MCMC Results
Converting Parameters to Data Frame
The results$Parameters object contains the MCMC chain of
all model parameters. To create trace plots and posterior distributions,
we use the mcmc_to_dataframe() function to convert this
nested list structure into a tidy data frame format.
# Convert parameters to data frame using package function
param_df <- mcmc_to_dataframe(results)
# Display first few rows
head(param_df)
#> iteration insitu_uncolonized insitu_colonized surv_test_uncol_neg
#> 1 1 0.4649756 0.53502440 1e-10
#> 2 2 0.8145916 0.18540844 1e-10
#> 3 3 0.1541333 0.84586666 1e-10
#> 4 4 0.9707602 0.02923979 1e-10
#> 5 5 0.8663606 0.13363938 1e-10
#> 6 6 0.4665449 0.53345514 1e-10
#> surv_test_col_neg surv_test_uncol_pos surv_test_col_pos clin_test_uncol
#> 1 0.8520047 1e-10 0.7868844 0.5
#> 2 0.7822836 1e-10 0.9999999 0.5
#> 3 0.8599913 1e-10 0.6272905 0.5
#> 4 0.8281964 1e-10 0.9998167 0.5
#> 5 0.7709993 1e-10 0.4185874 0.5
#> 6 0.8377648 1e-10 0.9998530 0.5
#> clin_test_col clin_rate_uncol clin_rate_col outunit_acquisition
#> 1 0.5 1 1 0.001181636
#> 2 0.5 1 1 0.001181636
#> 3 0.5 1 1 0.001181636
#> 4 0.5 1 1 0.001181636
#> 5 0.5 1 1 0.001181636
#> 6 0.5 1 1 0.001181636
#> outunit_clearance inunit_base inunit_time inunit_mass inunit_freq
#> 1 0.01 0.0008107566 1 1 1
#> 2 0.01 0.0006954064 1 1 1
#> 3 0.01 0.0006984290 1 1 1
#> 4 0.01 0.0007232861 1 1 1
#> 5 0.01 0.0008540253 1 1 1
#> 6 0.01 0.0008682528 1 1 1
#> inunit_colabx inunit_susabx inunit_susever inunit_clr inunit_clrAbx
#> 1 1 1 1 0.008567051 1
#> 2 1 1 1 0.007480784 1
#> 3 1 1 1 0.006921057 1
#> 4 1 1 1 0.006731989 1
#> 5 1 1 1 0.005782676 1
#> 6 1 1 1 0.005212212 1
#> inunit_clrEver abxrate_uncolonized abxrate_colonized loglikelihood
#> 1 1 1 1 -13020.82
#> 2 1 1 1 -13024.24
#> 3 1 1 1 -13033.93
#> 4 1 1 1 -13007.50
#> 5 1 1 1 -13025.07
#> 6 1 1 1 -13054.66Trace Plots
Trace plots show the evolution of parameters across MCMC iterations, helping to assess convergence.
library(ggplot2)
library(tidyr)
# Select key parameters for trace plots
trace_params <- param_df[, c("iteration", "insitu_colonized", "surv_test_col_pos",
"outunit_acquisition", "inunit_base",
"abxrate_colonized", "loglikelihood")]
# Convert to long format
trace_long <- pivot_longer(trace_params,
cols = -iteration,
names_to = "parameter",
values_to = "value")
# Create trace plots
ggplot(trace_long, aes(x = iteration, y = value)) +
geom_line() +
facet_wrap(~parameter, scales = "free_y", ncol = 2) +
theme_minimal() +
labs(title = "MCMC Trace Plots",
x = "Iteration",
y = "Parameter Value")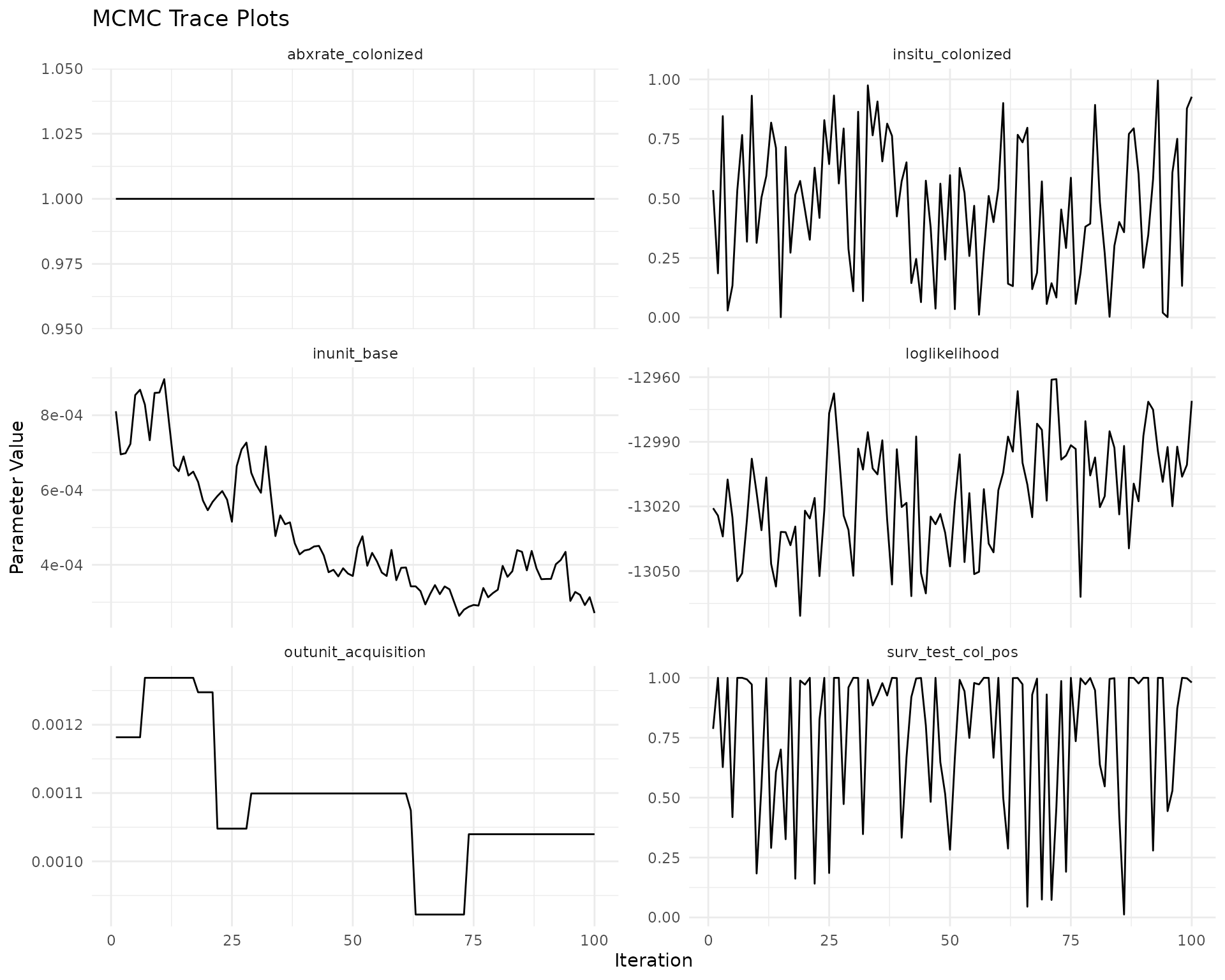
Posterior Distributions
Posterior distributions show the estimated distribution of each parameter after the MCMC sampling.
# Remove burn-in if needed (here we already set nburn in the MCMC call)
# For demonstration, let's use all samples since nburn=0 was specified
# Create density plots for posterior distributions
ggplot(trace_long, aes(x = value)) +
geom_density(fill = "steelblue", alpha = 0.5) +
geom_vline(aes(xintercept = mean(value, na.rm = TRUE)),
color = "red", linetype = "dashed") +
facet_wrap(~parameter, scales = "free", ncol = 2) +
theme_minimal() +
labs(title = "Posterior Distributions",
subtitle = "Red dashed line shows posterior mean",
x = "Parameter Value",
y = "Density")
Summary Statistics
# Calculate summary statistics for each parameter
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
summary_stats <- trace_long %>%
group_by(parameter) %>%
summarise(
mean = mean(value, na.rm = TRUE),
median = median(value, na.rm = TRUE),
sd = sd(value, na.rm = TRUE),
q025 = quantile(value, 0.025, na.rm = TRUE),
q975 = quantile(value, 0.975, na.rm = TRUE),
.groups = "drop"
)
print(summary_stats)
#> # A tibble: 6 × 6
#> parameter mean median sd q025 q975
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 abxrate_colonized 1 1 0 1 e+0 1 e+0
#> 2 insitu_colonized 0.466 0.494 0.285 7.16e-3 9.32 e-1
#> 3 inunit_base 0.000480 0.000430 0.000167 2.84e-4 8.60 e-4
#> 4 loglikelihood -13014. -13015. 25.5 -1.31e+4 -1.30 e+4
#> 5 outunit_acquisition 0.00109 0.00110 0.0000951 9.22e-4 1.27 e-3
#> 6 surv_test_col_pos 0.766 0.966 0.304 7.37e-2 1.000e+0